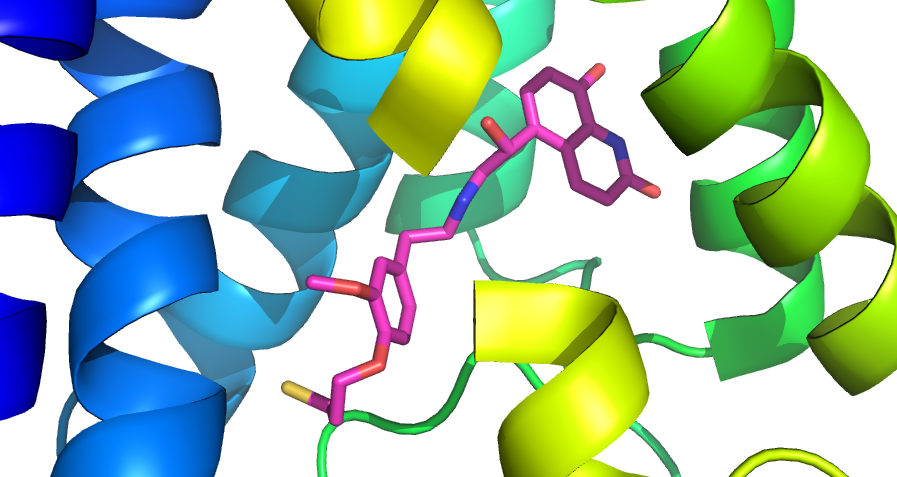
Such models may be used to filter/prioritise compound collections sets for allosteric compounds, for particular screening campaigns or for compound collection enrichment.
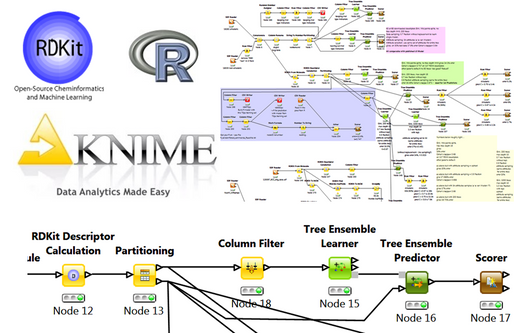
The Knime generated models also provide a prediction confidence value for each molecule. In a test case, a small number of compounds from a commercial screening collection were found to be present in the ChEMBL data set.
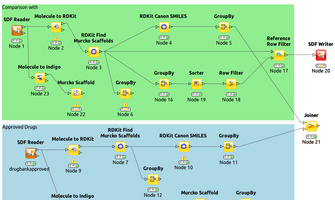
Models built using the same strategy - van Westen classification, RDKit descriptors, Knime machine learning nodes, 70:30 training:test set split - but with overlap compounds excluded, produced robust models showing a very low total prediction error of 14% for compounds with a prediction confidence >0.67